 |
|
|
||||
|
|
|
|
||||
|
|
有機化学のための 量子化学計算入門
|

|

|
||
| ハートリー-フォック法 【量子化学】DFT (密度汎関数理論) の基礎(1) (2024/6/28公開,約17分間) | DFTとコーン-シャム方程式 【量子化学】DFT (密度汎関数理論) の基礎(2) (2024/6/28公開,約15分間) |

|
|
| DFTに分子軌道が登場するワ
ケ 【量子化学】DFT (密度汎関数理論) の基礎-おまけ (2024/7/5公開,約11分間) → 補足情報 |
◎ 演習問題解答例 (4章〜10章;zip圧縮ファイル。2023/1/13更新)
◎ 8章付録(Reaction plus 基本チュートリアル) (©HPCシステムズ株式会社;pdfファイル)
◎ はじめに
◎ 索引 (以上pdfファイル)
【書評】化学ポータルサイト「Chem-Station」(2022/6/10掲載)
◎ 正誤表 (pdfファイル)
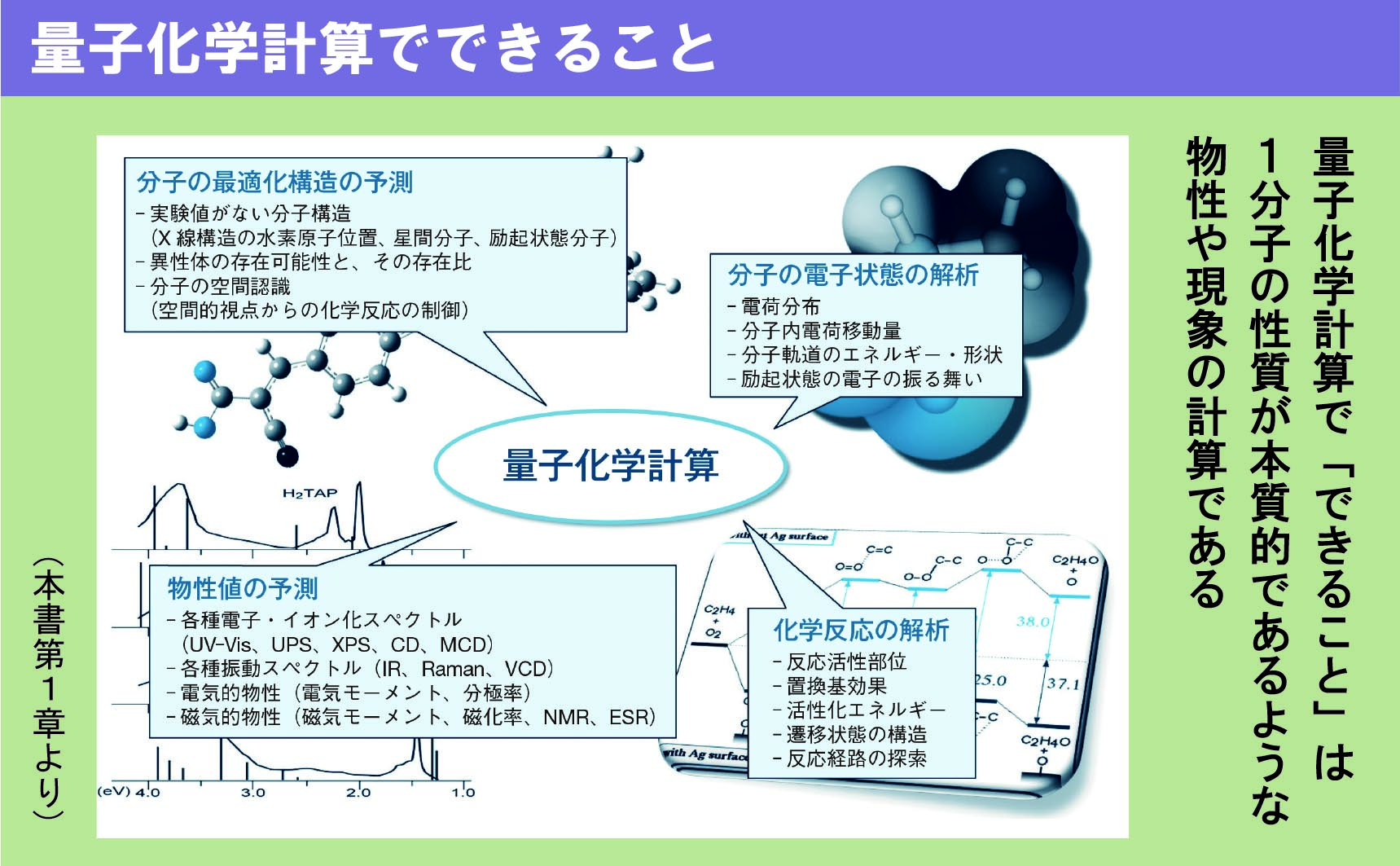
1.量子化学計算で何ができるか?
2.計算実行のための環境づくり
3.計算手法と基底関数
4.Gaussianの基本的な使い方
5.構造最適化
6.分子軌道
7.基底状態の物性
8.化学反応メカニズム
9.開殻系の取り扱い
10.励起状態の物性
11.計算を有効活用するためのヒント
はじめに-1/はじめに-2 (pdfファイル)
1.量子化学計算で何ができるか?
1.1 シミュレーション手法の概観・特徴比較
1.1.1 研究対象のスケールとシミュレーション手法の選択
1.1.2 化学系シミュレーション手法の特徴比較
1.2 量子化学計算でできること
1.3 量子化学計算の応用事例
1.4 おわりに
参考文献
2.計算実行のための環境づくり
2.1 量子化学計算ソフト
2.1.1 Gaussianの概要・特徴
2.1.2 その他の量子化学計算ソフトの概要・特徴
2.2 ビューアーソフト
2.2.1 GaussViewの概要・特徴
2.2.2 その他のビューアーソフトの概要・特徴
2.3 支援計算ソフト
2.4 計算機・クラウド環境
2.4.1 オンプレミス運用とクラウド運用
2.4.2 岡崎スパコンの利用
2.5 計算時間と並列効率
2.6 おわりに
参考文献
3.計算手法と基底関数
3.1 とりあえずお勧めの方法を知りたい人へ
3.2 量子化学計算の簡単な理論的背景
3.2.1 シュレーディンガー方程式
3.2.2 シュレーディンガー方程式の近似
3.2.3 計算手法と基底関数
3.3 計算手法
3.3.1 ハートリー -フォック法
3.3.2 電子相関と電子相関理論
3.3.3 密度汎関数理論
3.4 基底関数
3.4.1 基本基底関数
3.4.2 追加基底関数
3.4.3 有効内殻ポテンシャル
3.5 計算条件の選択
3.5.1 DFT汎関数の選択指針
3.5.2 基底関数の選択指針
参考文献
4.Gaussianの基本的な使い方
4.1 GaussianとGaussViewの基礎知識
4.2 Gaussianでの計算の流れ
4.3 Gaussian入力ファイルの構成
4.3.1 Link 0コマンド
4.3.2 ルートセクション
4.3.3 分子指定セクション
4.4 GaussViewによる入力ファイルの作成
4.4.1 一般的な分子構造の構築例
4.4.2 分子構造構築時でのその他の便利な機能
4.4.3 入力ファイルの作成
4.5 テキストエディタによる入力ファイルの編集
4.6 Gaussianジョブの実行
4.6.1 GaussViewからの実行
4.6.2 UNIX/Linuxでの実行
4.7 一般的な注意事項
4.7.1 入力ファイルのチェック
4.7.2 計算開始直後のチェック
演習問題
5.構造最適化
5.1 構造最適化とは
5.1.1 構造最適化の原理
5.1.2 初期構造選択の重要性
5.1.3 極小点の確認:振動解析
5.2 Gaussianによる構造最適化の手順
5.2.1 入力ファイルの作成と出力ファイルの見方
5.2.2 部分構造最適化
5.2.3 構造最適化後の振動解析
5.3 構造最適化のポイント
5.3.1 初期構造について
5.3.2 元素ごとに異なる基底関数を与える場合の構造最適化
5.3.3 構造最適化の進捗状況の確認
5.4 構造最適化の実例
演習問題
参考文献
6.分子軌道
6.1 分子軌道とは
6.1.1 原子価結合法と分子軌道法の違い
6.1.2 分子軌道の成り立ち
6.1.3 分子軌道間の相互作用
6.2 HOMOとLUMOの性質と利用法
6.2.1 HOMOとLUMOの性質
6.2.2 フロンティア軌道と反応性
6.3 Gaussianによる分子軌道計算の手順
6.3.1 入力ファイルの作成と出力ファイルの見方
6.3.2 分子軌道の可視化
6.4 応用事例
演習問題
参考文献
7.基底状態の物性
7.1 電荷分布
7.1.1 Mulliken密度解析
7.1.2 自然密度解析
7.1.3 電荷分布、双極子モーメント、静電ポテンシャルの可視化
7.2 IR・ラマンスペクトル
7.2.1 調和近似
7.2.2 IRとラマンスペクトルの計算
7.3 NMRスペクトル
7.3.1 化学シフトの計算
7.3.2 NMR計算の応用例:NICS
7.4 溶媒モデル
7.4.1 溶媒モデルの概要
7.4.2 PCM法および関連の方法
7.4.3 Gaussianでの溶媒効果の計算例
演習問題
参考文献
8.化学反応メカニズム
8.1 反応経路解析の基礎知識
8.1.1 基本用語
8.1.2 反応経路解析の一般的な手順
8.2 反応経路の最適化
8.2.1 SCAN法
8.2.2 NEB法
8.3 遷移状態の探索
8.3.1 通常法
8.3.2 QST2/QST3法
8.4 遷移状態の妥当性評価
8.4.1 振動解析
8.4.2 IRC解析
8.5 遷移状態構造最適化が上手くいかない時
演習問題
9.開殻系の取り扱い
9.1 開殻系とは
9.1.1 閉殻系と開殻系
9.1.2 制限法と非制限法
9.2 Gaussianによる計算の手順
9.2.1 二重項の計算
9.2.2 三重項と開殻一重項の計算:SCF解の安定性
9.2.3 初期軌道の組み換え
9.3 応用事例
9.3.1 2価の銅の2核錯体の交換相互作用
9.3.2 オリゴチオフェンラジカルカチオンπダイマー
演習問題
参考文献
10.励起状態の物性
10.1 励起状態計算の基礎知識
10.1.1 励起状態とは?
10.1.2 電子状態・分子軌道・電子配置
10.1.3 吸光・発光と分子構造の関係
10.2 励起状態の計算方法
10.2.1 励起状態計算手法の概観
10.2.2 TD-DFT法
10.2.3 基底関数の選択指針
10.3 Gaussian計算の手順・結果閲覧
10.3.1 1点計算
10.3.2 構造最適化計算
10.3.3 Gaussian計算上の注意
演習問題
参考文献
11.計算を有効活用するためのヒント
11.1 計算の3つの有効活用法
11.1.1 想定した仮説の妥当性を計算によって検証する
11.1.2 計算結果から新たな仮説を立てる
11.1.3 実験の代わりとして計算する
11.2 計算研究における3つのポイント
11.2.1 計算対象のモデル化
11.2.2 「計算可能量」への落とし込み
11.2.3 計算結果の妥当性評価
11.3 実験と計算それぞれの利点を意識する
11.4 おわりに
参考文献
索引 (以上pdfファイル)
![]()
西長 亨
にしなが とおる
1967年 大阪府に生まれる。京都大学大学院工学研究科博士課程修了。日本学術振興会特別研究員、京都大学化学研究所助手、首都大学東京准教授を経て現職。専門分野は構造有機化学、有機機能材料化学。主な著書に『マテリアルサイエンス有機化学(第2版)』(共著、東京化学同人)、『基礎から学ぶ有機化学』(共著、朝倉書店)などがある。
本田 康
ほんだ やすし
1972年 三重県に生まれる。京都大学大学院工学研究科博士課程修了。日本学術振興会特別研究員、首都大学東京助教、量子化学研究協会研究所部門副長を経てHPCシステムズ株式会社入社、2021年より現職。Gaussianプログラム(Gaussian 03以降)の公式コントリビューター。専門分野は理論化学・量子化学。
(情報は初版刊行時のものから一部修正しています)
![]()

パソコンで考える
量子化学の基礎

量子化学 上巻

量子化学 下巻











自然科学書出版 裳華房 SHOKABO Co., Ltd.







{kind=link}